Пошаговая инструкция по запуску Gromacs
Перед тем, как приступить к работе с пакетом Gromacs на системе Blue Gene/P, рекомендуется выполнить данную пошаговую инструкцию для новых пользователей, чтобы убедиться, что ваше окружение правильно сконфигурировано, и что вам успешно удается запускать Gromacs-задачи на суперкомпьютере.
Данная пошаговая инструкция составлена на основе демонстрационного примера gmxdemo.
Обратите внимание, что
команда mdrun должна использоваться только для запуска расчетов на фронтэнде,
а команда mdrun_mpi предназначена для использования только на суперкомпьютере.
Молекулярная динамика
-
Выполните загрузку модуля
gromacs:module load gromacsЕсли команда выдает ошибки, проверьте свою конфигурацию окружения Modules.
-
Скопируйте тестовый пример в свой домашний каталог и перейдите в директорию, в которой находится файл с описанием пептида, поведение которого будет моделироваться (
cpeptide.pdb):cp -r /gpfs/opt/fen/gromacs/default/share/gromacs/tutor ~
cd ~/tutor/gmxdemo -
В рамках данного примера идет работа только с одним пептидом. Для того, чтобы команды были более наглядными, экспортируйте имя файла с его описанием (без расширения) как переменную окружения
MOL:export MOL=cpeptide(Далее предполагается, что все команды выполняются в рамках того же самого терминала. Если планируется использовать несколько терминальных сессий, экспорт переменной необходимо произвести в каждой из них.)
-
Получите gro- и top-файлы из pdb-файла:
pdb2gmx -f ${MOL}.pdb -o ${MOL}.gro -p ${MOL}.top(В ответ на запросы "Select the Force Field" и "Select the Water Model" введите "1" в обоих случаях.)
-
Задайте для пептида пространственную область, в которой будет выполняться моделирование:
editconf -f ${MOL}.gro -o ${MOL}.gro -d 0.5 -
"Растворите" пептид в воде:
genbox -cp ${MOL}.gro -cs -o ${MOL}_b4em.gro -p ${MOL}.top -
Задайте параметры для стадии минимизации энергии — создайте в текущем каталоге файл с именем
em.mdpследующего содержания:define = -DFLEX_SPC
constraints = none
integrator = steep
nsteps = 100
nstlist = 10
ns_type = grid
rlist = 1.0
rcoulomb = 1.0
rvdw = 1.0
;
; Energy minimizing stuff
;
emtol = 1000.0
emstep = 0.01и обработайте его препроцессором:
grompp -f em -c ${MOL}_b4em -p ${MOL} -o ${MOL}_em -
Выполните минимизацию энергии:
mdrun -s ${MOL}_em -o ${MOL}_em -c ${MOL}_b4pr -v -
Задайте параметры для стадии подготовки к моделированию, в которой все связи в пептиде жестко зафиксированы, — создайте в текущем каталоге файл с именем
pr.mdpследующего содержания:define = -DPOSRES
constraints = all-bonds
integrator = md
dt = 0.002 ; ps !
nsteps = 500 ; total 1.0 ps.
nstcomm = 1
nstxout = 10
nstvout = 1000
nstfout = 0
nstlog = 10
nstenergy = 10
nstlist = 10
ns_type = grid
rlist = 1.0
rcoulomb = 1.0
rvdw = 1.0
; Berendsen temperature coupling is on in two groups
Tcoupl = berendsen
tau_t = 0.1 0.1
tc-grps = protein sol
ref_t = 300 300
; Pressure coupling is not on
Pcoupl = no
tau_p = 0.5
compressibility = 4.5e-5
ref_p = 1.0
; Generate velocites is on at 300 K.
gen_vel = yes
gen_temp = 300.0
gen_seed = 173529и обработайте его препроцессором:
grompp -f pr -c ${MOL}_b4pr -r ${MOL}_b4pr -p ${MOL} -o ${MOL}_pr -
Проведите расчет в условиях, когда все связи в пептиде жестко зафиксированы:
mdrun -s ${MOL}_pr -o ${MOL}_pr -c ${MOL}_b4md -v -
Наконец, задайте параметры для моделирования динамики пептида в растворе воды — создайте в текущем каталоге файл с именем
md.mdpследующего содержания:constraints = all-bonds
integrator = md
dt = 0.002 ; ps !
nsteps = 5000 ; total 10.0 ps.
nstcomm = 1
nstxout = 5
nstvout = 0
nstfout = 0
nstlist = 10
ns_type = grid
rlist = 1.0
rcoulomb = 1.0
rvdw = 1.0
; Berendsen temperature coupling is on in two groups
Tcoupl = berendsen
tau_t = 0.1 0.1
tc-grps = protein sol
ref_t = 300 300
; Pressure coupling is not on
Pcoupl = no
tau_p = 0.5
compressibility = 4.5e-5
ref_p = 1.0
; Generate velocites is on at 300 K.
gen_vel = yes
gen_temp = 300.0
gen_seed = 173529и обработайте его препроцессором:
grompp -f md -c ${MOL}_b4md -p ${MOL} -o ${MOL}_md -
Запустите расчет на суперкомпьютере:
mpisubmit.bg -m vn -n 8 -w 00:15:00 `which mdrun_mpi` -- -s ${MOL}_md -o ${MOL}_md -c ${MOL}_after_md -vПрограмма запускается в режиме VN (
-m vn). Для данного расчета задействовано всего лишь 8 вычислительных узлов (-n 8), так как система, которая моделируется, является сравнительно небольшой. Время расчета ограничено пятнадцатью минутами (-w 00:15:00). Обратите внимание, что имя исполняемого файла указывается суперкомпьютеру как`which mdrun_mpi`(не забывайте обратные одинарные кавычки!). Параметры передаются программе после символов--. -
Журнал расчета сохранится в файлах
md.logиmdrun_mpi.*.err. Их содержание можно изучить, например, с помощью командtailилиless.
Визуализация расчета
Если на вашей рабочей станции установлен пакет VMD, тогда вы можете выполнить визуализацию проведенного расчета.
-
Скопируйте на свою рабочую станцию файлы
cpeptide_b4md.groиcpeptide_md.trr, полученные в результате расчета. -
Запустите программу
vmd. -
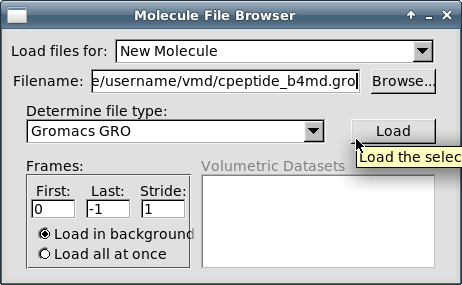
Выберите пункт меню "File - New molecule":
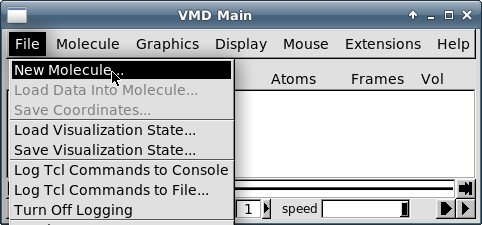
-
В открывшемся окне "Molecule File Browser" нажмите кнопку "Browse":
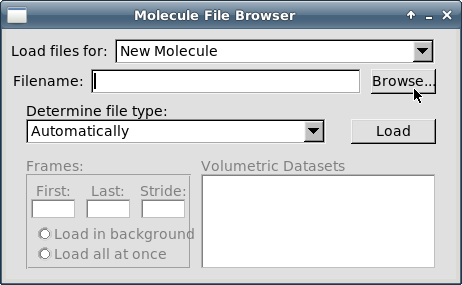
-
Чтобы загрузить описание молекулы, выберите файл
cpeptide_b4md.groи кликните на кнопке "OK":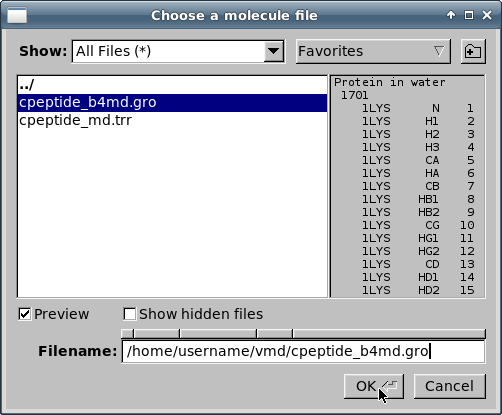
-
Кликните на кнопке "Load":
В окне визуализации вы увидите изображение примерно следующего вида:

-
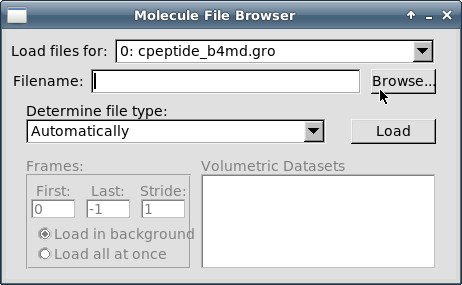
Теперь необходимо загрузить информацию о траекториях. Проверьте, чтобы в поле "Load files for:" было указано "0: cpeptide_b4md.gro". Кликните на кнопке "Browse":
-
Чтобы загрузить траектории, выберите файл
cpeptide_md.trrи кликните на кнопке "OK":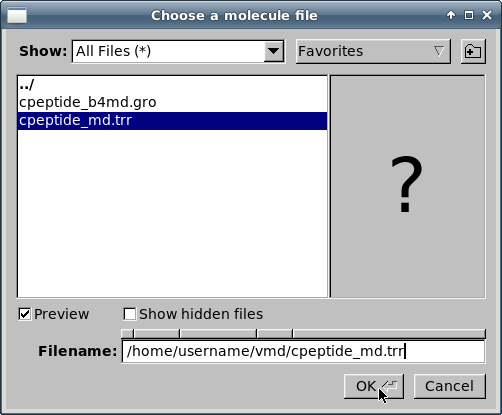
-
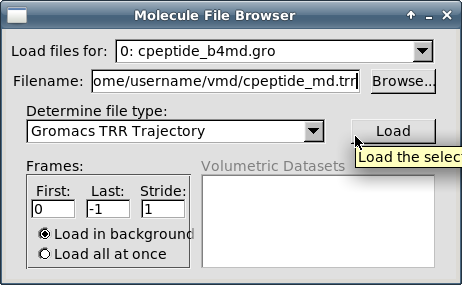
Кликните на кнопке "Load":
-
Закройте окно "Molecule File Browser".
Перейдите к пункту меню "Graphics - Representations":
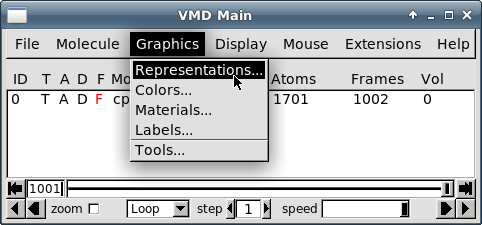
-
В окне "Graphical Representations" на вкладке "Draw style" введите "all not water" в поле "Selected atoms" и укажите "Licorice" в качестве "Drawing method":
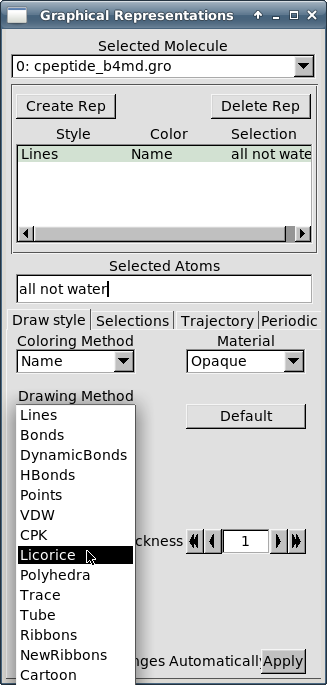
-
В том же окне "Graphical Representations" на вкладке "Trajectory" доведите значение параметра "Trajectory Smoothing Windows Size" до значения "5":
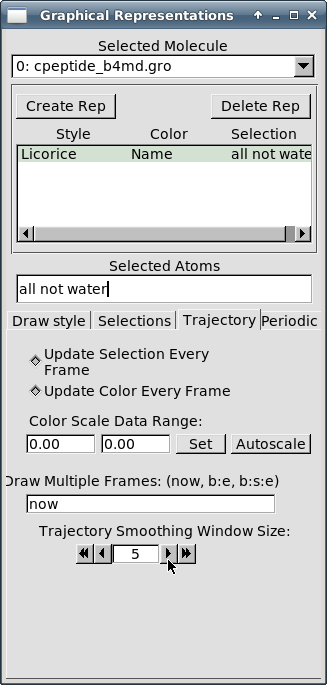
-
В главном окне кликните на расположенной в правом нижнем углу кнопке, означающей "воспроизведение":
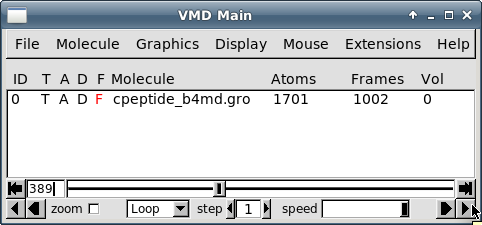
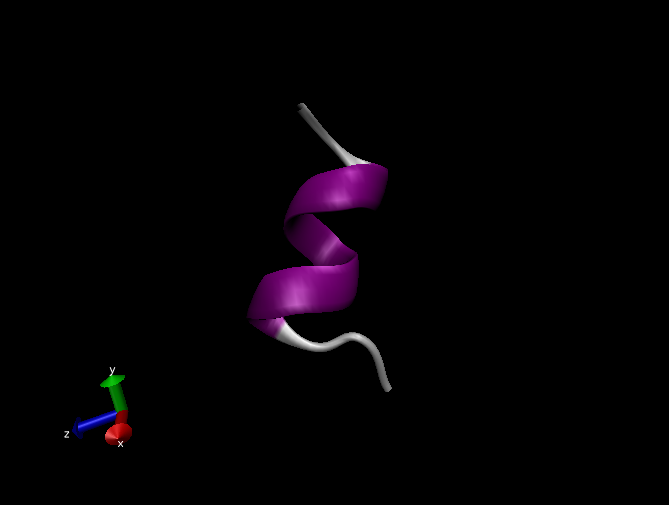
В окне визуализации вы увидите анимацию динамики пептида, который будет иметь примерно следующий вид:
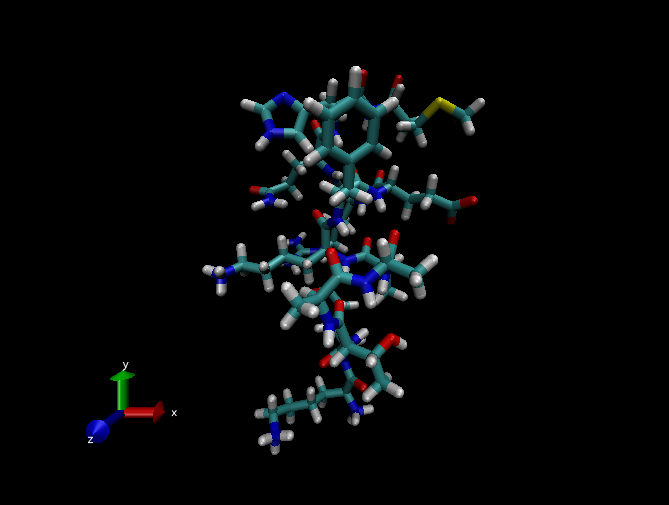
-
Если же в окне "Graphical Representations" на вкладке "Draw style" указать "Secondary Structure" в качестве "Coloring method" и "NewCartoon" в качестве "Drawing method", то картинка будет иметь следующий вид: